
Development of a Population Genomics Analysis Pipeline (PGAP) to Monitor Genetic Change at Neutral and Adaptive Genes
Summary:
Novel, well-evaluated computation methods can greatly advanced conservation and population genetics. Unfortunately, the statistical performance of many methods is unknown when dealing with vast amounts of multi-locus data and with data from natural population. Much of our research has focused on 'bridging the gap' between theory and application of statistical methods and estimators. This is becoming more difficult as genetic data sets are increasing in size and complexity faster than methods of data analysis can increase in speed, efficiency, and reliability.
We use simulations and empirical evaluations the power, accuracy, and precision of a new and existing computational methods and software designed to detect genetic change and molecular adaptation in natural populations. This will allow us to understand interactions among mechanisms underlying microevolutionary change (genetic drift, gene flow, and selection) and provide the scientific community with the means to conduct population genomics research as efficiently and effectively as possible.
With collaborators, we develop and evaluate Approximate Bayesian methods to estimate and monitor effective population size and to identify adaptive molecular variation. The methods will be applied to existing and new SNP data on domestic and wild fish and wildlife. The methods and software will allow researchers to evaluate the power and reliability of genetic monitoring methods to detect change in genetic parameters using simulated or real genetic marker data sets, and to address new questions not previously feasible.
Related projects:- Software for simulating complex demography and population genetic scenarios
- Software for simulating spatially explicit landscape genetics scenarios
- Effective population size and loss of genetic variation in large mammals: Effects of age structure, population fluctuations, and mating system.
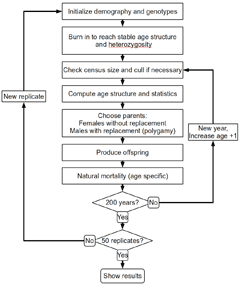
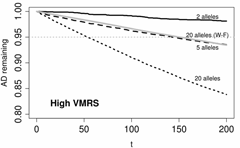
There is increasing need to estimate the effective population size (Ne) and rates of loss of genetic variation in natural populations, especially of large mammals and fish that are becoming increasingly isolated around the world. We calculated Ne and rates of loss of heterozygosity and allelic diversity in bison (Bos bison) from Yellowstone National Park using computer simulations of age structured populations with detailed demographic data on age- and sex-specific vital rates. We used a new computer program (NewAge; right) to simulate stable or fluctuating populations (lambda from 1.02 to 1.12), under a range of culling strategies (random, young, or adults only), a wide range of variance in male reproductive success, and for loci with allelic richness ranging from 2 to 20 alleles per locus.
Collaborators:
T. Antao, E. Landguth, T. Cozart, A. Pérez-Figueroa, A. M. Beaumont, A. Beja-Pereira, P. England, K. McKelvey, D. Tallmon, M. Schwartz, F. Allendorf, R. Waples, and others...
Publications:
- Antao, T., A. Beja-Pereira, and G. Luikart. 2007. modeler4simcoal2: A user-friendly, extensible modeler of demography and linked loci for coalescent simulations. Bioinformatics, 23:1848-1850.
- Antao, T., R.J. Lopes, A.Beja-Pereira, G. Luikart. 2008. LOSITAN: A workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinformatics, 9:323.
- Antao, T., A. Pérez-Figueroa, and G. Luikart. 2010. Detecting population declines: High power of genetic monitoring using effective population size estimators. Evolutionary Applications. 4: 144–154.
- Antao, T., I.M. Hastings, G. Luikart, M.J. Donnelly. Estimating effective population size in disease vectors: a critical assessment of applications and performance. In review. Berthier, P., M. A. Beaumont, J-M. Cornuet, and G. Luikart. 2002. Likelihood-based estimation of the effective population size using temporal changes in allele frequencies: a genealogical approach. Genetics, 160:741-751.
- England, P.R., G. Luikart, and R.S. Waples. 2010. Early detection of population fragmentation using linkage disequilibrium estimation of effective population size. Conservation Genetics, 11:2425-2430.
- England, P.R., J-M. Cornuet, P. Berthier, D.A. Tallmon and G. Luikart. 2006. Estimating effective population size from linkage disequilibrium: severe bias using small samples. Conservation Genetics, 7:303-308.
- Landguth, E.L., C.C. Muhlfeld, and G. Luikart. CDFISH: an individual-based, spatially-explicit, landscape genetics simulator for aquatic species in complex riverscapes. In review.
- Luikart, G., N. Ryman, D.A. Tallmon, M.K. Schwartz, and F.W. Allendorf. 2010. Estimating census and effective population sizes: Increasing usefulness of genetic methods. Invited Review, Conservation Genetics. 11: 355-373.
- Luikart, G., W. Sherwin, B. Steele, and F.W. Allendorf. 1998. Usefulness of molecular markers for detecting population bottlenecks via monitoring genetic change. Molecular Ecology, 7:963-974.
- Manel, S., F. Berthoud, E. Bellemain, M. Gaudeul, G. Luikart, J.E. Swenson, L.P. Waits, and P. Taberlet. 2007. A new individual-based spatial approach for identifying genetic discontinuities in natural populations: example application in brown bears. Molecular Ecology, 16:2031-2043.
- Pérez-Figueroa, A., R. Wallen, T. Antao, J. Coombs, F. Gardipee, M.K. Schwartz, F.W. Allendorf, and G. Luikart. Conservation of genetic variation in large mammals: effects of age structure, population fluctuations, and mating system in Yellowstone bison. In review.
- Tallmon, D., A. Koyuk, G. Luikart, and M. Beaumont. 2008. OneSamp: a program to estimate effective population size using approximate Bayesian computation. Molecular Ecology Resources, 8:299-301.
- Tallmon, D., G. Luikart, and M.A. Beaumont. 2004. Comparative evaluation of a new effective population size estimator based on approximate Bayesian summary statistics. Genetics, 167: 977-988.